
Introduction
Organic compounds are prevalent in lives of modern societies, and one of the most basic and most fundamental organic compounds is benzene. Not only it is widespread in nature because it is one of the natural constituents in crude oil, but it is also heavily used in industry. For example, benzene is added to gasoline at 1-2% by volume to prevent engine knocking[1], and it is also frequently used as a precursor for industrial synthesis of more complex organic compounds. However, the properties that contributed to benzene’s advantage in industrial use are also what contributed to its toxicity and prevented it from use in consumer product. Benzene is lipid soluble and highly volatile at room temperature, and that enables approximately 50% of inhaled benzene to be rapidly absorbed through the lungs[2]. From there, benzene is easily distributed throughout the body and accumulates in various organs including bone marrow and liver[2]. Prolonged exposures have been frequently associated with increased risks in numerous diseases like leukemia or cancer[3].
Given its toxicity, metabolism of benzene, both in body and in nature, are especially important. In nature, oxidative degradation of benzene by hydroxyl attack in the atmosphere turns benzene into phenol (hydroxybenzene), catechol (1,2-dihydroxybenzene), and quinol (1,4-dihydroxybenzene)[2]. Surprisingly, metabolism in human body also produces the same byproducts as metabolites, and they can be excreted through urine[1].
Therefore, both benzene and its immediate derivative phenol pose fascinating targets to study. Benefited from their relatively simple structures, computational methods like the PySCF package can be used to calculate their properties. For this study, I focused on bond stretching and bond rotation, and I investigated their effect on potential energy and molecular orbital of these two molecules. From the results of this study, I hope to provide a peek into the vast world of computational chemistry and create the steppingstones to computational analyses of more complex molecules.
Results
Starting with the highly symmetrical benzene, the properties of its C-H bond will be studied, including the effect of both bond stretching and rotation on potential energy.
▼ Molecular structure of benzene
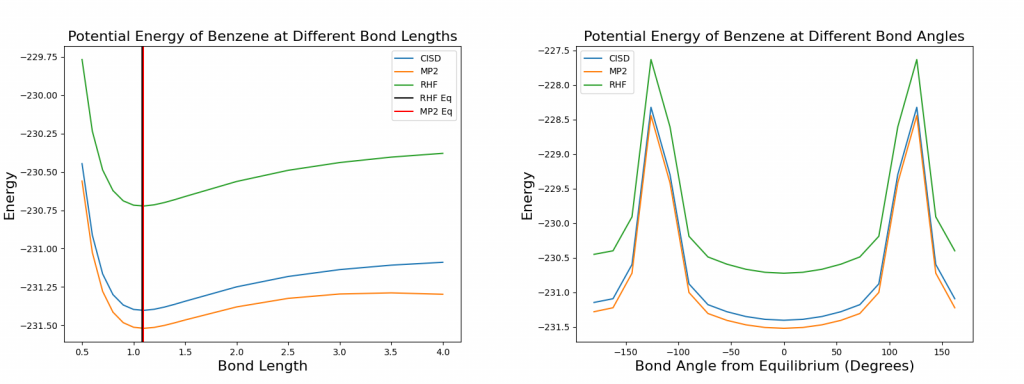
At about the same C1-H bond length, the minimum potential energy is reached for all three electronic structure methods, although the minimum energy varies. Hartree Fock produces the highest minimum potential energy while MP2 produced the lowest minimum energy. Comparing with results provided in CCCBDB, which are also marked on the plot, where equilibrium bond length is 1.0822 for RHF and 1.0952 for MP2. Without more precise calculations, it is hard to confirm if the results match exactly, but the values do fall into the right range as shown on the plot.
For the C2-C1-H bond rotation, it is shown that the equilibrium C2-C1-H bond angle from CCCBDB, which is 120 degrees for all methods, has the minimum potential energy. Given the symmetry of benzene, it is also observed that -120- and +120-degrees bond rotation will cause the H atom to collide with the carbon ring, which produced the spikes on the potential energy diagram. Once again, all three methods produced very similar equilibrium values for bond angles, but the exact potential energy at such bond angle still varies from one method to another. MP2 still has the lowest energy, while RHF still produces the highest energy.
From the potential energy surface (PES) diagrams, bond rotation has weaker impact on the molecular orbital than bond stretching does. Whereas bond rotation only changes the orientation of molecular orbitals with minimal distortion, different C-H bond lengths create drastically different PES, as shown in the appendix.
Since phenol is not completely symmetrical, there are a few bonds worth looking at. While the C-O bond stretching is unique and interesting to look at, its rotation is not vastly different from the C-C-H bond rotation in benzene. Therefore, a more interesting C-O-H bond rotation is investigated, along with its O-H bond stretching.
▼ Molecular structure of phenol
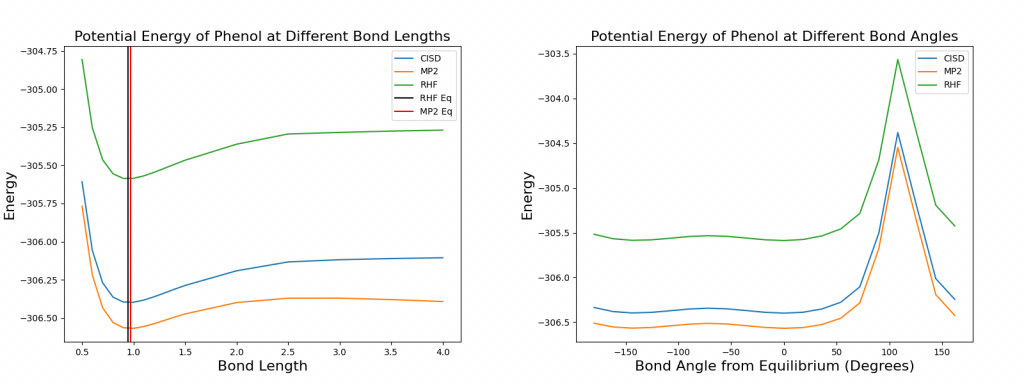
From the plot above, it is observed that the equilibrium bond length is very similar for all three methods. Without more precise calculations, it cannot be confirmed that the values match with those provided by CCCBDB exactly, which has the equilibrium bond length at 0.9452 for RHF and 0.9682 for MP2. However, the equilibrium values of these two methods do fall into the right range as shown in the plot above. In addition, the potential energy flattens a lot faster than benzene’s C-H bond stretching. Beside the difference in electronegativity, phenol’s unstable tautomer might also help stabilizing the molecule, since the tautomerization is most likely to happen when the bond stretches far.
In terms of the C1-O-H bond rotation, equilibrium bond angles are 110.284 degrees for RHF and 107.223 degrees for MP2, according to CCCBDB. There is an interesting trend shown in the plot above that in addition to the equilibrium bond angle provided by the database, there is another bond angle that has similar potential energy, and it is at roughly -144 degrees from the equilibrium. The explanation is straightforward from looking at the molecular structure below. Without numbering the closest carbon to the hydrogen, the two structures seem identical. In fact, the symmetry and nature of the conjugated monocyclic planar system provides that hydrogen the freedom to choose whichever side it wants to be, since these two positions are energetically equivalent. Therefore, two minima are observed in the plot above, and a maximum around the bond angles that will cause collision of atoms.
▼Two energetically equivalent phenol configurations of the O-H bond
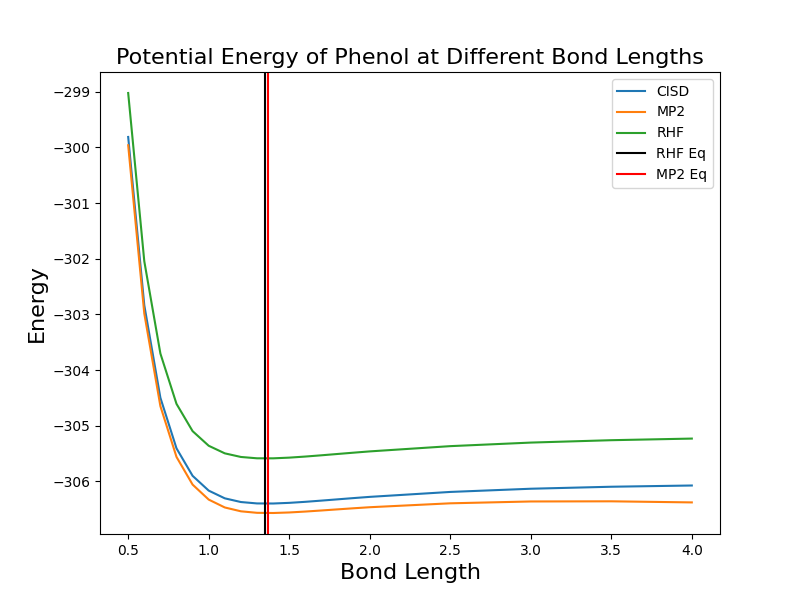
As for C1-O bond stretching, it had a larger impact on both potential energy and molecular orbital of phenol, especially when bond length is short. The equilibrium bond length of C1-O is 1.3506 for RHF and 1.3701 for MP2, and they again fall into the appropriate range as shown on the plot. The energy fluctuation also had a larger scale than previous plot did.
Contrary to bond rotation, it is observed that even stretching the O-H bond can create significant distortion on molecular orbital of the whole system, let alone stretching the C1-O bond. In addition, with the similarity in structures, it can also be interesting to compare the PES of pulling off a hydroxyl group from phenol with that of pulling off just one hydrogen atom from benzene. Due to the electronegativity of oxygen, C1-O bond stretching distorts the PES more prominently as shown in the appendix.
Appendix
1. Benzene Bond Stretching PES
2. Benzene Bond Rotation PES
3. Phenol CO Bond Stretching PES
4. Phenol Bond Stretching PES
5. Phenol Bond Rotation PES
[1] J.R. Kuykendall, in Encyclopedia of Ecology, 2008
[2] C. Barton, in Encyclopedia of Toxicology (Third Edition), 2014
[3] Erika L. Abel, John DiGiovanni, in The Molecular Basis of Cancer (Fourth Edition), 2015